Medical Device Regulations (EU and UK)
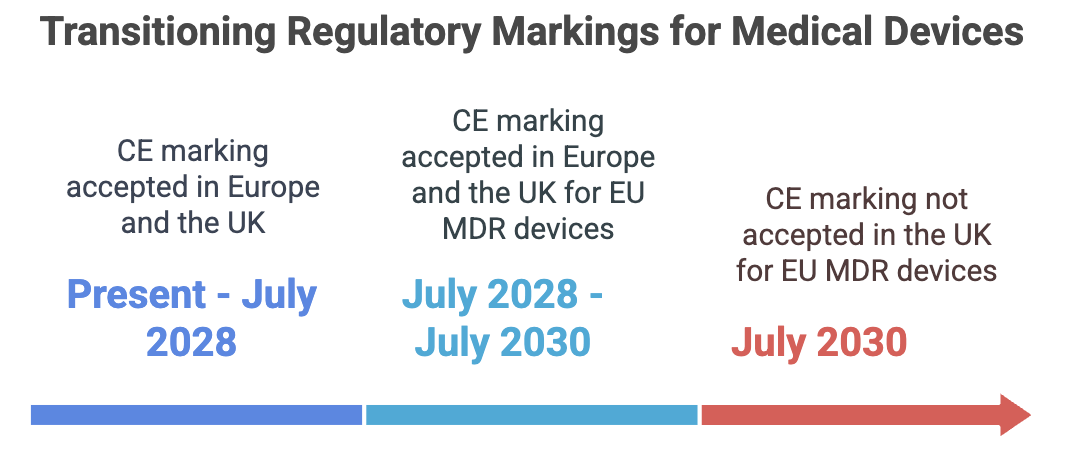
Medical devices must comply with specific regulatory frameworks before they can be marketed. In the European Union (EU), devices require a CE marking, indicating conformity with EU safety, health, and environmental requirements. Following Brexit, the United Kingdom (UK) introduced the UK Conformity Assessed (UKCA) marking for products sold in Great Britain (England, Wales, and Scotland). However, the UK continues to accept CE-marked devices during a transition period, the specifics of which are outlined below.
CE and UKCA Marking Transition Periods
Technical Documentation Requirements
Both CE and UKCA markings require manufacturers to maintain a comprehensive technical file demonstrating compliance with relevant regulations. While the core components of the technical file are similar, there are specific requirements unique to each marking:
- UKCA Technical File:
- Must include evidence of conformity with the UK Medical Devices Regulations 2002 (as amended). This involves detailed device descriptions, design and manufacturing information, risk assessments, and clinical evaluations.
- CE Technical File:
- Should demonstrate compliance with the EU Medical Device Regulation (MDR) or In Vitro Diagnostic Regulation (IVDR), including similar documentation tailored to EU-specific requirements.
Key Considerations for Manufacturers
- Conformity Assessment:
- Higher-risk devices (e.g., Class IIa, IIb, III) require assessment by a notified body (for CE marking) or an approved body (for UKCA marking). Manufacturers must engage with the appropriate body based on their target market.
- Labeling:
- Devices must bear the appropriate marking (CE or UKCA) and, where applicable, the identification number of the notified or approved body involved in the conformity assessment.
- Market Surveillance:
- Post-market surveillance obligations remain consistent across both jurisdictions, requiring manufacturers to monitor device performance and report adverse events.
Manufacturers aiming to market medical devices in both the EU and UK must navigate the nuances of CE and UKCA markings, ensuring adherence to the respective regulatory requirements and timelines. Staying informed about ongoing regulatory developments is crucial for maintaining compliance and market access.
References
- European Commission. (2017). Regulation (EU) 2017/745 on medical devices. Official Journal of the European Union. Retrieved from https://eur-lex.europa.eu/eli/reg/2017/745/oj
- Medicines and Healthcare products Regulatory Agency (MHRA). (n.d.). Regulating medical devices in the UK. GOV.UK. Retrieved from https://www.gov.uk/guidance/regulating-medical-devices-in-the-uk
- European Commission. (n.d.). New regulations: Medical devices and in vitro diagnostic medical devices. Retrieved from https://health.ec.europa.eu/medical-devices-sector/new-regulations_enPublic Health+1Wikipedia+1
- MHRA. (2024). MHRA publishes revised roadmap of regulatory framework for medical devices. Retrieved from https://www.emergobyul.com/news/mhra-publishes-revised-roadmap-future-regulatory-framework-medical-devicesEmergo by UL
- Avania. (2024). Medical device regulation changes in the UK. Retrieved from https://www.avaniaclinical.com/blog/medical-device-regulation-in-the-uk/Avania
- TÜV SÜD. (n.d.). The Medical Device Regulation (MDR). Retrieved from https://www.tuvsud.com/en-us/resource-centre/infographics/the-new-medical-device-regulationTÜV SÜD
- Baker McKenzie. (2024). MHRA roadmap for future UK medical device legislation. Retrieved from https://healthcarelifesciences.bakermckenzie.com/2024/01/16/mhra-roadmap-for-future-uk-medical-device-legislation/Healthcare & Life Sciences Blog